
Ted Kalbfleisch
- Lab Website
- Scholars@UK
- Member of the UK Genetics and Genomics Research Group
- Slides from CCS/RCD Seminar on 2/13/2024
The Challenges and Opportunities of Reference Free Genomic Data Analysis

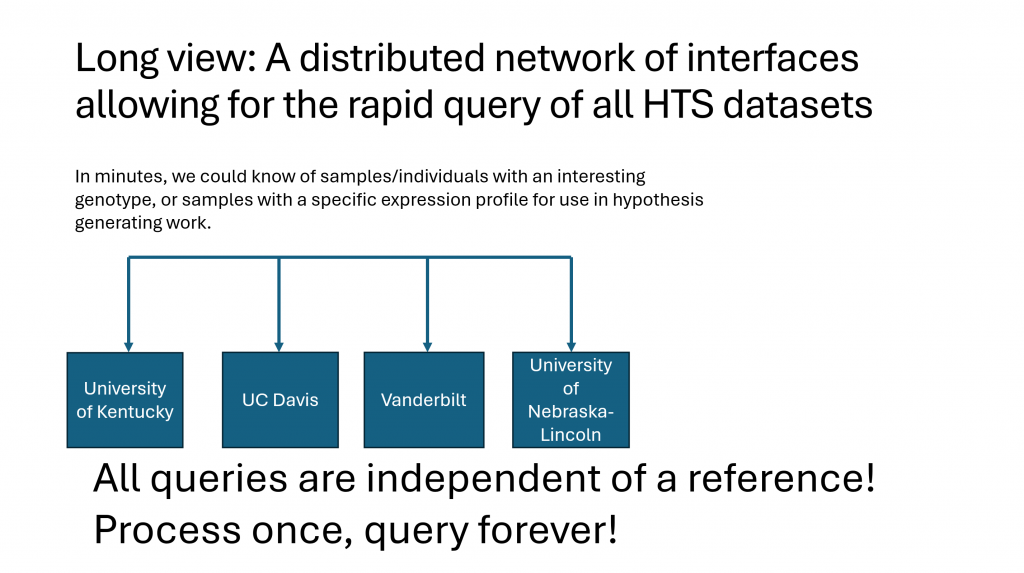
Since its inception, the cost of generating high quality genomic sequence has plummeted toward zero. Pipelines that perform primary analyses and genome assembly have scaled well with available computing power and have rarely been a rate limiting step for genomic analyses. Historically, a species has had a single reference genome which has been well annotated for gene structure and epigenetic marks. Sequence data for any animals from that or a closely related species are mapped to that reference for analysis in that curated genomic context. The advent of inexpensive long, and ultralong sequencing technologies have made the assembly of phased, nearly gapless genomes routine. The main advantages are that one can study genomes fully phased, as they are in nature, and large structural variants in ways that have been impossible previously. This has created a new paradigm where each of these new genomes will need to be analyzed in a context not of a reference genome, but rather in the larger context of all that is known. The co-emergence of this challenge and artificial intelligence creates a tremendous opportunity. With the insights of computer science colleagues, we can now begin to ponder the data structures that will be most amenable to analysis by AI technologies, as well as the metadata that include both molecular, and macroscopic phenotype data, and how these datasets could be readily accessed and analyzed in a distributed network of both computing and storage.
Supported by grants from NIFA, University of Texas Health Sciences Center at Houston, University of Arizona